Supplementary information
In this section you will find the materials and methods used for synthesizing the different fluorophores, for assembling and amplifying the DNA, transcribing it to RNA, and for collecting the fluorescence spectra. You will also find the supplementary figures and the designed sequences.Sequences
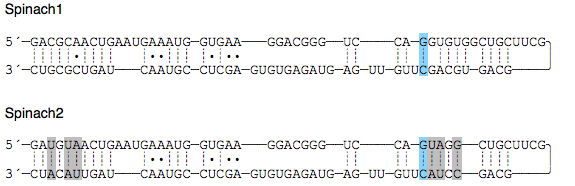

Polymerase Chain Reactions
All strands for PCR were bought at Integrated DNA Technologies. The components dNTP, 10x Buffer and Taq polymerase were acquired from Invitrogen – Life Technologies.To assemble DNA strands longer than 100 base pairs (bp), assembly PCR was used. To amplify the DNA sequences shorter than 100 bp or after assembly PCR, regular PCR was used. All PCR products were quality controlled on a 1% agarose gel.
Assembly PCR
The assembly PCR was carried out by splitting the DNA sequences into three fragments with an overhang of approximately 20 nucleotides (nt) with a melting temperature of approximately 55°C. First, fragment (1+2) assembles and during the next cycle fragment (3) assembles onto this fragment assembling all three fragments into one.
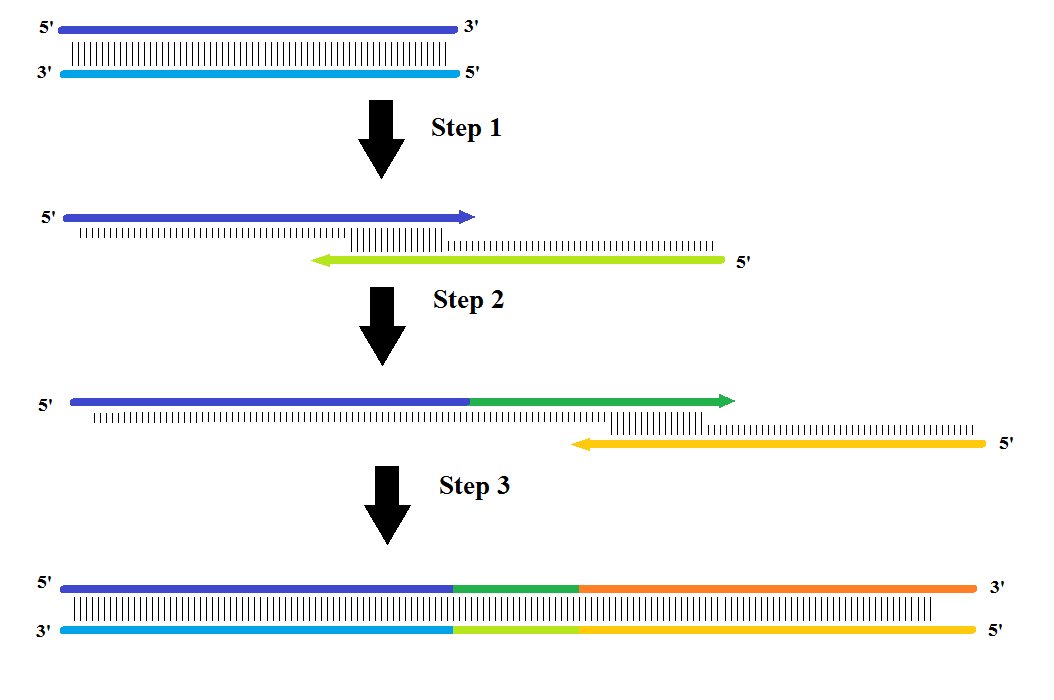
For the assembly PCR a mixture of 10 μL 10x Buffer, 0.8 μL 100 mM dNTP, 3 μL 50 mM MgCl2 and 75.69 μL H2O was made. The three sequences were then added, but to make sure the (1+2) fragments didn’t anneal with its complementary strand instead of fragment (3), the fragments (1) and (3) were added in 500x excess. Therefore 5μL of fragment (1) and (3) were added and 0.01 μL of fragment (2). Just before running the 30-cycle PCR, 0.5 μL Taq DNA polymerase was added. The program was initiated for 3 minutes at 94°C, then 30 cycles of: denaturation at 94°C for 30 sec., annealing at 50°C/52°C for 45 sec. and elongation at 72°C for 90 sec. At the end it elongated at 72°C for 10 minutes.
Full length PCR
For amplifying the assembly PCR, a full PCR was made. It consisted of 10 μL 10x Buffer, 0.8 μL 100 mM dNTP, 3 μL 50 mM MgCl2, 80.2 μL H2O, 0.5 μL of the forward primer, 0.5 μL of the reverse primer, 5 μL of the assembly PCR product and 0.5 μL Taq DNA polymerase. The PCR program was the exact same as for the assembly PCR.1% Agarose gel
1g agarose was dissolved in 100 mL 0.5x TBE by heating. After cooling to about 60°C, 7 μL SYBR Safe was added for staining. 0.5x TBE was used for buffer. The samples loaded on the gel was made of 2 μL DNA sample, and 1 μL native buffer (1 mL glycerol and 10 μg Sybr Gold), and one well in the agarose gel was loaded with 5 μL 100 bp DNA ladder from Invitrogen. The gels ran for 100 minutes at 100 Volt.Transcription
The transcription mixture consisted of 5 μL 10x Transcription Buffer, 5 μL 10 mM DTT, 20 μL DNA template, 15 μL 10 mM NTP mix, 4 μL H2O, 1 μL T7 RNA polymerase (1 μg/μL), giving a total volume of 50 μL. The mixture was added to a PCR tube and heated at 37°C for 4 hours.8% Polyacrylamide gel
An 8% polyacrylamide gel consisted of 29 mL Urea concentrate, 5 mL TBE(10x), 16 mL Urea diluent, 0.5 mL APS and 16 μL TEMED, giving a total volume of 50 mL. It was used for purifying the RNA with subsequent EtOH precipitation and was loaded with 100 µL RNA per well.The gel was prerun at 600 V for 15 minutes before loading RNA samples with RNA loading buffer (0.6 mL of 0.5 M EDTA, 700 μL of a mixture of formamide, xylencyanol and bromophenol blue supplied by H. C. Høiberg and 13.7 mL formamide). It was mixed 1:1 with RNA samples.
Precipitation
EtOH precipitation was used to extract RNA from the 8% polyacrylamide gel. The bands were cut out and left overnight in 300μL extraction buffer (0.3 M NaAc, 0.25 mM EDTA) on a shaker at 4°C. The next day the samples were centrifuged at 17.000 rpm at room temperature for 15 minutes, the supernatant was transferred to a new tube and mixed with 750 µL 96% EtOH. After vortexing, the samples were put on dry ice (-80°C) for 15 minutes and centrifuged again at 17.000 rpm at 4°C for 15 minutes. The supernatant was discarded, and 100-200 µL 70% EtOH was added, and centrifuged at 4°C for 15 minutes. The supernatant was again discarded and the pellets were dried in a fumehood and dissolved in 50 µL water.
For measuring the concentrations of the purified RNA transcripts, the NanoDrop 1000 spectrophotometer was used.
In vivo transcription
This is all the preparations and experimental protocols needed for in vivo transcription of the constructs.Making BL21 DE3 electrocompetent
1 l LB media, 250 ml 10% glycerol and 50 microcentrifuge tubes were autoclaved for 30 minutes at 121°C with 22 psi. The BL21DE3 cells were grown overnight in 1l LB media with 50 µg µl-1 kanamycin at 37°C. The media was then centrifuged at 4000 rpm at 4°C for 30 minutes. The supernatant was removed and the pellet was resuspended in 100 ml 10% glycerol. It was then centrifuged one more time at 4000 rpm at 4°C for 30 minutes and then washed. The washing was done by removing the supernatant, adding 100 ml 10 % glycerol and then centrifuging once more at 4000 rpm at 4°C for 30 minutes. The supernatant was removed and the pellet was resuspended in 50 ml 10% glycerol after which it was aliquoted to the microcentrifuge tubes and frozen on dry ice before immediately being put in a -80°C freezer. All samples and equipment used were kept on ice or in a cold room.LB media
10 g tryptone, 10 g NaCl and 5 g yeast extract was mixed and dissolved in 1 l Milli-Q water. The solution was sterilized by autoclaving for 30 minutes at 121°C. It was kept at room temperature to check for contamination.LB agar medium
10 g tryptone, 10 g NaCl, 5 g yeast extract and 15 g agar was mixed and dissolved in 1 l Milli-Q water. The solution was then sterilized by autoclaving for 30 minutes at 121°C. The media was cooled to 65°C and 50 µg µl-1 kanamycin was added. It was then poured into 5 cm petri dishes.
Expression of Spinach RNA
The Spinach plasmids were cleaved with XhoI to make sure transcription was terminated at the right place. This was done because the T7-terminator does not work as well in vitro as in vivo. 15 µl nuclease-free water, 2 µl 10x FastDigest® buffer, 2 µl DNA and 1 µl FastDigest® XhoI were mixed and incubated for 20 minutes at 37°C. Afterwards the enzyme was deactivated by elevating the temperature to 80°C for 5 minutes. 4 µl 10x Transcription buffer, 4 µl DTT, 2 µl NT mix and 4 µl T7 polymerase were added to the digestion mixture. The transcription mixture was incubated for 14 hours at 37°C. It was then separated on a 10% polyacrylamide gel, at 600 volts, 40 volt/cm, 40 watts for 1 h 45 m. The band with the RNA was visualized with UV-light and extracted from the gel into 600 µl of RNase-free water by electroelution in MIDI GeBaFlex-tubes. 60 µl of NaAc acetate was added, and the solution was split into two microcentrifuge tubes, where 1155 µl of EtOH 96% was added (temperature - 20°C). The samples were put on dry ice for 15 minutes and subsequently centrifuged in the cold room for 30 minutes at 17,000 g. The supernatant was then removed and 200 µl of 70% EtOH (temperature - 20°C) was added. The samples were centrifuged as before and almost all of the supernatant was removed. It was allowed to dry and was then resuspended in RNase-free water.
Concentrations were measured on a NanoDropTM 1000 with RNase-free water as a blank.
Plasmid amplification
50 µl of electrocompetent DH5α cells were transformed with 4 µl of plasmid in 0.2 cm electroporation cuvettes. The cuvettes were placed on ice before use. The mixture of cells and plasmid was pulsed at 2.5 kV on the Biorad Genepulser with settings set to 0.2 cm cuvettes. Immediately after pulsing, 1 ml LB media was added, and the resuspended cells were incubated for 1 h (1.5 h if it was a ligated plasmid) at 37°C with 225 rpm shaking. The cells were then plated on LB plates with 50 µg µl-1 kanamycin. The plasmid was then extracted by performing a maxiprep using the NucleoBond® PC500 kit from Macherey-Nagel according to manufacturer's instructions. The plasmids were sequenced at GATC-biotech for confirmation. A quick screening was performed before sending the plasmid to sequencing. A plasmid profile was made utilising electrophoresis on a 1% agarose gel at 11 volt/cm. Some lanes had showed cleavage by XhoI and/or BglII.
Ligation
To create the pET28c-positive plasmid (the spinach gene inside the pET28c vector), PCR assembled Spinach DNA and the pET28c-SAM were both cleaved with BglII and XhoI (FastDigest® Thermo Scientific) according to the manufacturer's instructions, incubation of 40 minutes at 37°C. They were then subjected to electrophoresis on a 1% agarose gel at 11 V/cm and stained with SybrSafe. SybrSafe was chosen as a stain because of the easy visualization under UV-light but it was not needed. Under UV-light the band with the DNA was cut and the DNA was extracted by using the Freeze 'N Squeeze kit from Quatum Prep. The vector DNA and insert (assembly DNA) was then ligated using T4 DNA ligase from Thermo scientific (EL0016) according to manufacturer's instructions with freshly made 10x DNA ligase buffer. The only change from the protocol was incubation at 22°C, which was done for an hour instead of ten minutes. 5 µl from the ligation mixture was then dialyzed using Millipore® MF-Millipore™ DNA Filter Paper for a half hour. This step was needed to ensure that the salt concentration was low enough for usage in the Biorad Gene-pulser. The plasmid was then ready for transformation.
M9 5x salt solution
96.5 g Na2HPO4 · H20, 15 g KH2PO4, 2.5 NaCl and 5 g NH4Cl was mixed and dissolved in a final volume of 1 l Milli-Q water. The solution was then autoclaved for 30 min, at 22 psi and 121°C.
M9 medium
200 ml 5x M9 salt solution, 2 ml 1 M MgSO4, 100 µl 1 M CaCl2 was mixed, the pH was adjusted to 6.0 and a final volume adjusted to 1 l with Milli-Q water. In one of the mediums 2 mg ml-1 glucose was added, in another medium 2 mg ml-1 glucose and 2 mg ml-1 methionine was added. The final medium was sterilized by vacuum filtration through a 0.22-µm-pore filter.
Student's t-test
A Student's t-test was performed to test significance between the different samples. For all tests two tails were chosen along with double sample with different variance, since this fitted the data best.
Fluorescence measurements
All fluorescence measurements were made on the FluoroMax-3 (Inc 1 s, slit length 5 nm (Orthogonality tests) or 10 nm (SAM sensor construct), integration time 0.05 s, 37°C).
Measurements were made with the following excitation wavelength and emission range:
Spinach + DFHBI: Excitation 460 nm, emission range 480 to 600 nm.
Mango + TO3: Excitation 610 nm, emission range 620 to 800 nm.
17-3 / 2-4 / 3-6 + DMHBI: Excitation 400 nm, emission range 410 to 600 nm.
11-3 + DMABI: Excitation 480 nm, emission range 500 to 600 nm.
6-8 + 2-HBI: Excitation 400 nm, emission range 500 to 600 nm.
DFHBI, DMHBI, 2-HBI, DMABI and TO3 were dissolved in a selection buffer with 40 mM HEPES, 125 mM KCl, 1 mM MgCl2 and 2 µM fluorophore. RNA was added for a final concentration of 0.26 µM.
Microscope imaging of fluorescent cells
To express the Spinach-RNA, BL21 DE3 E.coli cells were transformed with the plasmid, as described above. The cells were plated and incubated at 37°C overnight. A sample from a colony for the positive control, negative control and Spinach SAM aptamer were incubated with 1 ml M9 media for 26 h. The reason for extensive time was the lack of nutrient in M9 media. Four hours before the measurement, the samples were induced with 1 mM IPTG. One hour before the measurement, a 19 µl aliquot was taken out and 1 µl 4 mM of DFHBI stock was added for a 200 µM final concentration of DFHBI. 20 minutes before the measurement, the aliquots were put on a microscope slide with a cover slip on top and sealed with nail polish to prevent evaporation. The fluorescence was detected with a confocal microscope LSM 700 (Zeiss) with excitation at 488 nm 10 mW. The images were then analysed using Image J software where the mean fluorescent intensities per cell were calculated.
Synthesis of DFHBI and DFHBI analogs
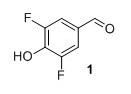
3,5-difluoro-4-hydroxy benzaldehyde (1) To a round-bottomed flask 2,6-difluorophenol (1.50 g, 11.5 mmol) and hexamethylenetetramine (1.60 g, 11.5 mol) in TFA (10 mL) were added. The mixture heated at reflux under argon overnight. The solution was then cooled to room temperature and the solvent was evaporated in vacuo. The crude residue was mixed with CH2Cl2 and washed with NaHCO3. The aqueous layers were acidified to pH=1 with concentrated HCl. The solution was then extracted with CH2Cl2, dried over MgSO4 and the solvent was evaporated to dryness in vacuo. The product was passed through a silica plug, washed with pentane after which remaining solvent was evaporated in vacuo. Compound 1 was obtained as a white solid (1.17 g, 64%). Rf 0.38 (1:2 EtOAc/pentane). mp (uncorr.) 119.7-122.4 °C. 1H NMR (400 MHz, CDCl3) δH (ppm) 9.82 (t, 1H), 7.5 (m, 2H), 5.85 (s, 1H). 13C NMR (100 MHz, CDCl3) δC (ppm) 189.2, 152.0 (dd, J=256 Hz and 6 Hz), 139.0 (t, J=16 Hz), 128.3 (t, J=6 Hz), 113.5 (dd, J=15 Hz and 7 Hz). 19F NMR (377 MHz, CDCl3) δF (ppm) -132.9. HRMS (ESI) m/z [M+H]+ calc. for C7H5O2F2 159.0252, found 159.0247.

4-acetate-3,5-difluorobenzylidene oxazolone (2) 1 To a round-bottomed flask 1 (0.1998 g, 1.26 mmol), N-acetylglycine (0.1497 g, 1.28 mmol), NaOAc (0.1037 g, 1.26 mmol) and Ac2O (1.0 mL) were added. The mixture stirred at 110 °C for 2.5 h and was then cooled to room temperature. Cold EtOH (4 mL) was added and the reaction cooled at 4°C overnight. The resulting solid was washed with cold EtOH, pentane after which remaining solvent was evaporated in vacuo. Compound 2 was obtained as a yellow solid (0.2764 g, 78%). Rf 0.57 (100% CH2Cl2). mp (uncorr.) 145.5-147.0 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 7.75 (d, J=8.4 Hz, 2H), 6.96 (s, 1H), 2.43 (s, 3H), 2.39 (s, 3H); 13C NMR (100 MHz, CDCl3): δC (ppm) 167.8, 167.2, 167.1, 155.2 (dd, J=250 Hz and 47 Hz), 134.6, 131.9 (t, J=9 Hz), 129.1 (t, J=17 Hz), 127.7 (t, J=3 Hz), 115.6 (dd, J=18 Hz and 6 Hz), 20.4, 16.1. 19F NMR (377 MHz, CDCl3) δF (ppm) -124.9. HRMS (ESI) m/z [M+H]+ calc. for C13H10NO4F2 282.0572, found 282.0569.
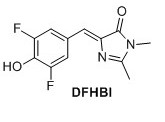
3,5-difluoro-4-hydroxy benzylidene imidazolinone (DFHBI) 1 A solution of 2 (0.030 g, 0.107 mmol) in EtOH (4.0 mL) was mixed with K2CO3 (0.023 g, 0.160 mmol) and CH3NH2 (0.05 mL). The mixture heated at reflux for 3 h. Upon cooling a precipitate formed, which was filtered and extracted with EtOAc and NaOAc (500 mM pH 3.0). The organic layers were separated, dried over MgSO4 and evaporated in vacuo. The crude compound was purified by flash chromatography (6:1 EtOAc/pentane with 1% formic acid). The title compound was obtained as a yellow solid (0.0135 g, 50%). Rf = 0.30 (6:1 EtOAc/pentane with 1% formic acid). 1H NMR (400 MHz, CD3OD): δH (ppm) 7.77 (dd, J=8 Hz and 1.6 Hz, 2H), 6.91 (s, 1H), 3.19 (s, 3H), 2.44 (s, 3H), missing OH proton due to deuterium exchange; 19F NMR (377 MHz, CD3OD) δF (ppm) -136.4. HRMS (ESI) m/z [M+H]+ calc. for C12H11F2N2O2F2 253.0783, found 253.0782. It was not possible to obtain a 13C NMR spectrum since the sample could not get concentrated enough.
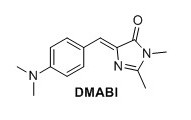
4-dimethylamine benzylidene imidazolinone (DMABI) 1 To a round-bottomed flask N-acetylglycine (2.06 g, 13.4 mmol), sodium acetate (1.12 g, 13.4 mmol), 4-dimethylaminobenzaldehyde (1.60 g, 13.4 mmol) and Ac2O (6.3 mL) were added. The mixture was stirred under argon at 60°C for 1 h and then refluxed overnight. The brown solid was dissolved in water, extracted with CH2Cl2, dried over MgSO4, and concentrated in vacuo. The obtained solid (0.260 g, 1.13 mmol) was mixed with CH2NH2 (0.25 mL, 40% in H2O), K2CO3 (0.077 g, 0.45 mmol) in EtOH (7.0 mL) and refluxed under argon overnight. The mixture was evaporated in vacuo, and the solid was purified by recrystallization from EtOH. The title compound was obtained as an orange solid (0.0349 g, 1%). Rf 0.20 (1:1 EtOAc/pentane). mp (uncorr.) 208.6-209.7°C. 1H NMR (400 MHz, CDCl3) δH (ppm); 8.05 (d, J=8.8 Hz, 2H), 7.08 (s, 1H), 6.71 (d, J=9.2 Hz, 2H), 3.18 (s, 3H), 3.06 (s, 6H), 2.36 (s, 3H) 13C NMR (100 MHz, CDCl3) δC (ppm) 175.6, 171.0 159.1, 134.2, 129.0, 122.3, 111.8, 40.3, 26.9, 20.6, 15.5. HRMS (ESI) m/z [M+H]+ calc. for C14H18N3O 244.1444, found 244.1447.
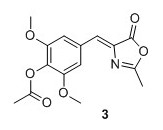
(Z)‐2,6‐dimethoxy‐4‐((2‐methyl‐5‐oxooxazol‐4(5H)‐ylidene)methyl)phenyl acetate (3) 1 Syringaldehyde (3.6696 g, 20.1 mmol), N-acetylglycine (2.47 g, 21.1 mmol) and sodium acetate (1.74 g, 21.2 mmol) were added to Ac2O (15.0 mL) and heated to 100°C. The solution was stirred at reflux for 16 hours. The solution obtained an orange color. Cold EtOH (20 mL) was added and the solution was stirred at 4°C for 16 hours. The yellow precipitate was collected by filtration and washed with cold EtOH, then pentane to yield 4.67 g, 76% of compound 3 as a yellow solid. 1H NMR (400 MHz, CDCl3) δH 7.42 (s, 2H), 7.05 (s, 1H), 3.88 (s, 6H), 2.40 (s, 3H), 2.35 (s, 3H). 13C NMR (100 MHz, DMSO‐d6) δ 167.9, 167.4, 166.9, 151.8 (2C), 132.6, 131.3, 130.3, 129.4, 108.9(2C), 56.1 (2C), 20.1, 15.6 . Mp(uncorr.)=168°C‐170°C HRMS (ESI) m/z [M + H]+ calcd. for C15H16NO6+, 306.0972, found 306.0974
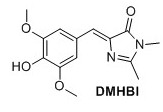
(Z)‐5‐(4‐hydroxy‐3,5‐dimethoxybenzylidene)‐2,3‐dimethyl‐3,5‐dihydro‐4H‐imidazol‐4‐one (DMHBI) 1 Compound 3 (1.12 g, 3.67 mmol), K2CO3 (705 mg, 5.10 mmol) and 40% aqueous MeNH2 (0.5 mL, 5.78 mmol) were added to EtOH (10 mL) and stirred for 3 hours at 75°C. The solution was cooled to room temperature and poured into 1:1 EtOAc:H2O and NaOAc (500 mM pH 3.0) and stirred briefly. The organic layer was separated. Addition of NaCl to the water phase caused an emulsion to form. The water phase was extracted with EtOAc (3 x 30 mL). The organic layers containing the emulsion were combined, dried over Na2SO4 and concentrated to yield 163 mg, 16 % of the title compound as a pale orange solid. 11H NMR (400 MHz, DMSO-d6) δH 7.61 (s, 2H), 6.88 (s, 1H), 3.78 (s, 6H), 3.08 (s, 3H), 2.32 (s, 3H). OH signal not observed. 13C NMR (100 MHz, DMSO-d6) δc 169.8, 161.8, 148.0(2C), 140.1, 136.1, 126.2, 123.6, 110.3(2C), 56.0 (2C), 26.2, 15.5 . Rf = 0.25(8:2 EtOAc/Pentane). Mp(uncorr.)=205°C(decomp) HRMS (ESI) m/z [M+H]+ calcd for C14H17N2O4+, 277.1183, found 277.1183. UV λmax= 382 nm
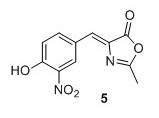
Z‐4‐(4‐acetoxy‐3‐nitro‐benzylidene)‐2‐methyl‐4H‐oxazol‐5‐one4 (5)4 To a mixture of 4‐hydroxy‐3‐nitro‐benzaldehyde (2.00 g, 12.0 mmol), N‐ acetylglycine (2.80 g, 23.9 mmol) and sodium acetate (1.96 g, 23.9 mmol) were added Ac2O (12.0 mL). The solution was heated to 100°C and stirred at reflux temperature for 16 hours obtaining an orange color. Cold EtOH (20 mL) was added and the solution was cooled to 0°C and stirred for 1 hour after which it was allowed to precipitate at 4°C for 16 hours. The yellow precipitate was filtered, washed with cold EtOH and pentane to yield 1.54 g, 44% of compound 5 as a yellow solid. 1 H NMR (400 MHz, CDCl3) δH 8.88 (d, J = 2.0 Hz, 1H), 8.29 (dd, J = 8.5, 2.0 Hz, 1H), 7.31 (d, J = 8.5 Hz, 1H), 7.08 (s, 1H), 2.44 (s, 3H), 2.39 (s, 3H). 13C NMR (100 MHz, CDCl3) δC 168.4, 168.2, 167.0, 145.2, 142.2, 137.5, 135.0, 132.1, 128.8, 126.8, 125.8, 20.9, 15.9. Mp(uncorr.)=164.8°C ‐166.5°C HRMS (ESI) m/z [M + H]+ calcd for C13H11N2O6 + , 291.0612, found 291.0609.
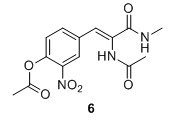
(Z)‐2‐acetamido‐3‐(4‐hydroxy‐3‐nitrophenyl)‐N‐methylacrylamide (6) Compound 5 (1.50 g, 5.17 mmol), K2CO3 (1.58 g, 11.43 mmol) and 33% MeNH2 in EtOH (2.6 mL, 20.7 mmol) were added to EtOH (15 mL) and stirred for 3 hours at 75 °C. The solution was poured into 1:1 EtOAc:H2O and NaOAc (500 mM pH 3.0) and a yellow brown emulsion formed in the organic phase. The layers were separated and the water phase extracted with EtOAc (3 x 30 mL). The organic layers were combined and dried over Na2SO4 and evaporated to yield 745 mg, 51 % of compound 6 as a pale orange solid 6H NMR (400 MHz, DMSO‐d6) δ 9.37 (s, 1H), 8.06 (d, J = 2.0 Hz, 1H), 7.94 (d, J = 4.6 Hz, 1H), 7.64 (dd, J = 8.8, 2.0 Hz, 1H), 7.10 (d, J = 8.8 Hz, 1H), 7.04 (s, 1H), 2.66 (d, J = 4.6 Hz, 3H), 2.03 (s, 3H). 13C NMR (100 MHz, DMSO‐d6) δ 169.3, 165.2, 153.4, 136.8, 136.0, 129.0, 126.1, 125.8, 124.5, 119.7, 26.3, 23.0. Mp(uncorr.)= 148°C‐149°C. HRMS (ESI) m/z [M + H]+ calcd. for C12H14N3O5+, 280.0928, found 280.0930.
(Z)‐5‐(4‐hydroxy‐3‐nitrobenzylidene)‐2,3‐dimethyl‐3,5‐dihydro‐4H‐imidazol‐4‐one (NBI) Compound 6 (745 mg, 2.67 mmol) was dissolved in pyridine (10.0 mL, 124 mmol) and heated to 115°C after which it was stirred for 24 hours.3 The solvent was removed in vacuo and the resulting oil was filtered through silica with EtOAc to yield 276 mg, 40% of the title compound as an orange solid. 1H NMR (400 MHz, DMSO‐d6) δH 11.53 (s, 1H), 8.78 (d, J = 2.0 Hz, 1H), 8.31 (dd, J = 8.8, 2.0 Hz, 1H), 7.16 (d, J = 8.8 Hz, 1H), 6.94 (s, 1H), 3.08 (s, 3H), 2.34 (s, 3H). 13C NMR (100 MHz, DMSO‐d6) δC 170.1, 164.9, 153.5, 138.8, 138.5, 137.7, 128.8, 126.1, 123.0, 119.8, 26.7, 15.9 . Rf=0.5 (EtOAc).Mp(uncorr.)= 188°C (decomp.). HRMS (ESI) m/z [M+H]+ calcd. for C12H12N3O4+, 262.0822 found 262.0824. UV λmax=367 nm
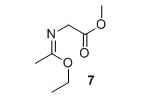
Methyl (E)‐2‐((1‐ethoxyethylidene)amino)acetate (7)2 Methyl glycinate hydrochloride (1.02 g, 8.09 mmol) and K2CO3 (1.12 g, 8.09 mmol) were suspended in Et2O (40 mL) and water (3.5 mL). Ethyl acetimidate hydrochloride (1.01 g, 8.09 mmol) was added and the suspension was stirred vigorously for 6 minutes. The organic layer was decanted off and the H2O phase was extracted with Et2O (30 mL). The organic layers were combined and dried over MgSO4. Solvent was removed in vacuo to yield 601 mg, 46% of compound 7 as a colorless liquid. 1H NMR (400 MHz, CDCl3) δH 4.06 (q, J = 7.1 Hz, 2H), 4.00 (s, 2H), 3.68 (s, 3H), 1.83 (s, 3H), 1.21 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δC 171.7, 164.8, 61.0, 51.9, 51.2, 15.3, 14.2. HRMS (ESI) m/z [M+H]+ calcd. for C7H14NO3 +, 160.0968 found 160.0971
N‐methyl‐1‐aryl‐methanimines2 An aromatic aldehyde (1.00 mmol) and 40% aqueous MeNH2 (95 µL, 1.10 mmol) were added to EtOH (1 mL) The reaction was allowed to stir for 12 hours. The solvent was removed in vacuo to yield the desired compound in near quantitative yield.
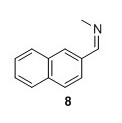
8: Yield 162 mg 96%.2 1H NMR (400 MHz, CDCl3) δ 8.43 (s, 1H), 8.05 – 7.93 (m, 2H), 7.92 – 7.80 (m, 3H), 7.57 – 7.47 (m, 2H), 3.58 (d, J=1.3 Hz 3H). 13C NMR (100 MHz, CDCl3) δ 162.7, 134.7, 134.0, 133.2, 129.8, 128.7, 128.6, 128.0, 127.2, 126.6, 123.7, 48.4 . Mp (uncorr.)=83°C‐84°C. HRMS (ESI) m/z [M+H]+ calcd for C12H12N+, 170.0964, found 170.0965.
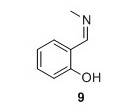
9: Yield 134 mg 99%. 1H NMR (400 MHz, CDCl3) δH 13.47 (s, 1H), 8.35 (d, J = 1.4 Hz, 1H), 7.30 (t, J = 8.5 Hz, 1H), 7.24 (d, J = 7.5 Hz, 1H), 6.95 (d, J = 8.5 Hz, 1H), 6.86 (t, J = 7.5 Hz, 1H), 3.49 (d, J = 1.4 Hz, 3H). 13C NMR (100 MHz, CDCl3) δC 166.3, 161.3, 132.1, 131.1, 121.5, 118.6, 117.2, 46.2. HRMS (ESI) m/z [M+H]+ calcd for C8H10NO+, 136.0757 found 136.0758.
General cycloaddition imidazolinone formation from compound 8 and 92 To the aldimine in EtOH (1 mL) freshly made 7 (1 eq) was added and the reaction was allowed to stir for 24 hours. The mixture was cooled to 0°C, suspended in pentane, filtered and washed with cold Et2O (2mL) and cold EtOH (1 mL) to yield the desired product.
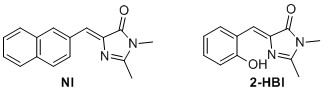
(Z)‐2,3‐dimethyl‐5‐(naphthalen‐2‐ylmethylene)‐3,5‐dihydro‐4H‐imidazol‐4‐one2 (NI) 2 The general cycloaddition imidazolinone formation procedure was used with 8 (0.5 mmol, 84.6 mg) to yield 8.90 mg 7% of the title compound as a yellow solid. 1 H NMR (400 MHz, CDCl3) δH 8.52‐8.32 (m, 2H), 8.02 – 7.72 (m, 3H), 7.56 – 7.42 (m, 2H), 7.26 (s, 1H), 3.21 (s, 3H), 2.42 (s, 3H). 13C NMR (100 MHz, CDCl3) δC 170.9, 162.6, 139.0, 134.1, 133.4, 133.3, 132.1, 129.1, 128.5, 128.3, 127.8, 127.6, 127.6, 126.5, 26.8, 15.9. Mp(uncorr.)=205°C (literature 192°C‐193°C) HRMS (ESI) m/z [M+H]+ calcd for C16H15N2O+, 251.1179, found 251.1182. UV λmax= 366 nm.
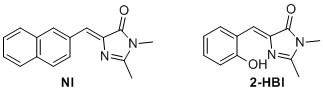
(Z)‐5‐(2‐hydroxybenzylidene)‐2,3‐dimethyl‐3,5‐dihydro‐4H‐imidazol‐4‐one (2-HBI)6 The general cyclodaddtion imidazolone procedure was used with 9 (0.7 mmol, 94.6 mg) to yield 115.06 mg, 76% of the title compound as a yellow solid. 1 H NMR (400 MHz, CDCl3) δH 13.75 (s, 1H), 7.34 (t, J=8.2 Hz 1H), 7.29 (d, J = 7.8 Hz, 1H), 7.16 (s, 1H), 6.95 (d, J = 8.2 Hz, 1H), 6.85 (t, J=7.8 Hz, 1H), 3.21 (s, 3H), 2.38 (s, 3H). 13C NMR (100 MHz, CDCl3) δC 168.1, 158.7, 157.6, 136.5, 134.3, 132.9, 130.4, 119.8, 119.6, 119.4, 26.9, 15.3. Rf=0.52 (8:2 EtOAc/pentane). Mp(uncorr.)=163‐164°C HRMS (ESI) m/z [M+H]+ calcd. for C12H13N2O2+, 217.0972, found 217.0975. UV λmax= 391 nm
Synthesis of Mango Fluorophores
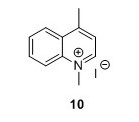
1,4-methylquinolinium iodide (10) 5 To a flask containing 1,4-dioxane (85 mL) were added 4-methylquinoline (6 mL, 45.4 mmol) and MeI (6 mL, 96.4 mmol). The solution was heated to 105°C. The solution gradually attained a brown through orange color and a yellow compound precipitated (and some darkbrown/black material was on the glass too). After 1.5 hours the reaction was allowed to cool to room temperature (RT). The solution was filtered and the filter cake was washed with Et2O (3 x 25 mL) and pentane ( 3 x 25 mL) to yield 10 g, 81% of compound 10 as a yellow solid. Rf=0.08 (EtOAc). 1H NMR (400 MHz, DMSO) δH 9.40 (d, J = 6.2, 1H), 8.52 (dt, J = 2.1, 4.5, 1H), 8.50 (d, J = 2.1, 1H), 8.31 – 8.22 (m, 1H), 8.10 – 8.01 (m, 2H), 4.59 (s, 3H), 3.00 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δC 158.0, 148.8, 137.5, 134.8, 129.5, 128.3, 126.6, 122.3, 119.4, 45.0, 19.5. HRMS (ESI) exact mass calculated for [M]+ C11H12IN+ requires m/z 158.0964 Found 158.0978
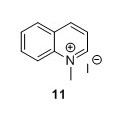
1-Methylquinolinium iodide (11)5 To a flask containing 1,4-dioxane (150 mL) were added MeI (10 mL, 161 mmol) and quinoline (9.15 mL, 77.4 mmol). The solution was heated to reflux and stirred for 1 h. The solution obtained an orange colour that gradually changed to red/brown. The solution was cooled to RT and the yellow precipitate was collected by filtration, mortared and washed with Et2O (3 x 50 mL) and pentane ( 3 x 50 mL) to yield 81%, 16.6 g of compound 11 as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.54 (d, J = 5.7 Hz, 1H), 9.30 (d, J = 8.4 Hz, 1H), 8.51 (dd, J = 12.0, 8.7 Hz, 2H), 8.32 – 8.24 (m, 1H), 8.18 (dd, J = 8.4, 5.7 Hz, 1H), 8.06 (t, J = 7.6 Hz, 1H), 4.65 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 150.1, 147.0, 138.3, 135.4, 130.3, 129.9, 129.1, 122.0, 119.1, 45.5.
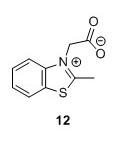
2-(2-Methylbenzo[d]thiazol-3-ium-3-yl)acetate (12)5 To a 100 mL round bottomed flask were added 2-methylbenzothiazole (1.7 mL, 13.40 mmol), bromoacetic acid (2.9098 g, 20.97 mmol) and toluene (30 mL). The mixture was heated to 111°C and stirred for 15 hours. The solution was filtered and washed with toluene (3 x 15 mL). A white grey solid with a pink tinge was isolated. Remaining solvent was removed in vacuo to yield 1.25 g, 45% of compound 12. 1H NMR (400 MHz, DMSO-d6) δH 8.53 (dd, J = 8.1, 0.7 Hz, 1H), 8.31 (d, J = 8.4 Hz, 1H), 7.93 – 7.84 (m, 1H), 7.84 – 7.77 (m, 1H), 5.80 (s, 2H), 3.23 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δC 179.0, 166.5, 141.0, 129.5, 128.5, 128.2, 124.8, 116.5, 49.9, 17.0. HRMS (ESI) [M + H]+ calc. m/z 208.0427, found 208.0435
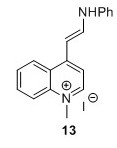
4-[(E)-2-Anilinoethenyl]-1-methylquinolinium iodide (13)5 To a flask containing EtOH (50 mL) were added 11 (10.5 g, 36.7 mmol, 1 eq) and N,N′-diphenylformamidine (14.2 g, 72.1 mmol, 2.0 equiv.). The solution was heated to 80°C after which it obtained a brown/red color and was stirred for 1.2 hours. The reaction was monitored by TLC analysis (EtOAc). A purple precipitate was isolated by filtration and washed with EtOH (3 x 25 mL) and dried in vacuo overnight. 1H-NMR analysis revealed impurities. Another round of washing with EtOH (3 x 50 mL) was performed after which the compound was dried in vacuo and finally subjected to lyophilisation for 48 hours. 14.75 g of crude compound was isolated and used in the next step without further purification. HRMS (ESI) exact mass calculated for [M+, C18H17IN2+] requires m/z 261.1386 Found 261.1402
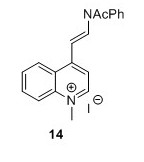
4-[(E)-2-(Acetylphenylamino)ethenyl]-1-methylquinolinium iodide (14)5 To a flask containing Ac2O (30 mL, 317 mmol, 17.6 eq) and pyridine (100 mL, 1.24 mol, 66 eq) was added 14 (7.0 g, 18.0 mmol, 1 eq). The solution turned lime green and was stirred for 20 hours. EtOAc (250 mL) was added and the solution was filtered and the resulting green precipitate was washed with EtOAc (3 x 75 mL) and suspended in CH2Cl2(40 mL). The suspension was filtered and the filtrate was concentrated in vacuo. The filtered compound was resuspended in CH2Cl2 (100 mL) and poured into Et2O (175 mL) after which the suspension was filtered. The resulting green solid was cryodessicated over night to yield 5.01 g, 32 % in two steps. 1H NMR (400 MHz, DMSO-d6) δH 9.17 (d, J = 6.6 Hz, 1H), 8.88 (d, J = 14 Hz, 1H), 8.36 (s, 1H), 8.34 (d, J = 2.3 Hz, 1H), 8.22 – 8.11 (m, 1H), 7.93 – 7.82 (m, 2H), 7.74 – 7.61 (m, 3H), 7.58 – 7.50 (m, 2H), 6.02 (d, J = 14.0 Hz, 1H), 4.48 (s, 3H), 2.09 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 169.7, 152.6, 147.5, 138.3, 137.9, 134.7, 130.5, 129.7, 129.1, 128.6, 125.3, 124.8, 119.4, 114.7, 102.7, 44.4, 40.1, 23.3.HRMS (ESI)[M+H+,C20H19N2O+] requires m/z 303.1492 found 303.1508
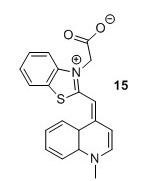
TO1 acetate (15)5 To a flask containing CH2Cl2 (12 mL) were added compound 12 (1.3 g, 1 eq), Et3N (2 mL, 3 eq) and compound 13 (1 g, 1 eq). The solution was stirred for 38 hours after which it was concentrated in vacuo. The resulting crude compound was then suspended in EtOH ( 8 mL) and Et2O (2 mL) for 4 days at 4 °C upon which a precipitate formed. The precipitate was isolated by filtration and redissolved in MeOH (8 mL) and H2O (2 mL) after which it was stored at 4 °C for 24 hours. The precipitate was isolated by filtration, suspended in acetone and stirred for 1 hour after which it was collected by filtration and washed with acetone (3 x 4 L) to yield 150 mg, 9% of the title compound as a crimson red solid. HRMS (ESI) [M+H+, C20H17N2O2S+] requires m/z 349.1005. Found 349.1008

TO3 acetate (16)5 Et3N (0.40 mL, 2.87 mmol, 0.5 eq) was added to a stirred solution of compound 15 (2.49 g, 5.79 mmol, 1 eq) and compound 13 (1.20 g, 5.79 mmol, 1 eq) in CH2Cl2( 40 mL). Upon addition the color of the solution changed from brown to green. The solution was left to stir for 22 hours after which it was concentrated in vacuo. The resulting solid was resuspended in CH2Cl2 (150 mL), filtered and washed acetone (3 x 10 mL). The resulting blue compound was dried in vacuo to yield 1.21 g of TO3 acetate as a regal blue solid. HRMS (ESI) Exact mass calculated for [M+H]+,C22H19N2O2S+ requires m/z 375.1162. Found 375.1165
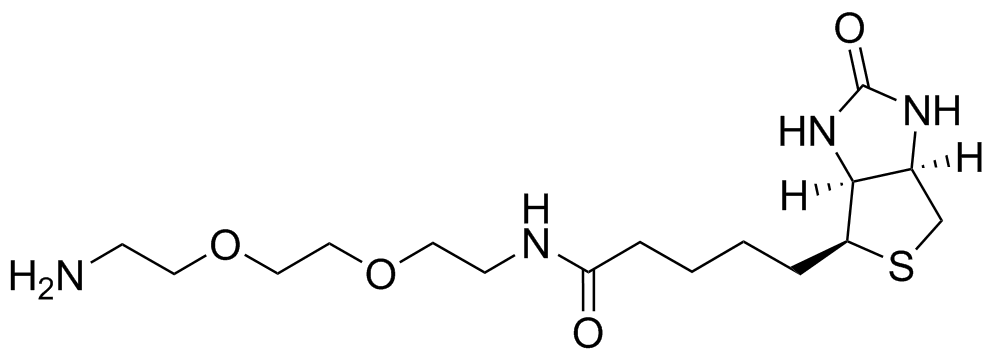
General procedure for EZ-link amine-PEG2 biotin formation: To a vial containing DMF (1 mL), 1,4-dioxane (1 mL) and H2O (0.5 mL) were added Biotin (125 mg) and HBTU (244 mg). The solution was stirred for 15 minutes and turned into a white suspension. The PEG 2 (4 eq) was added and the solution was stirred for another 15 minutes becoming colorless in the process. The solution was then lyophilised and the crude compound was used without further purification.
General procedure for TO1 & TO3 derivatives:5 To a vial containing DMF (250 uL) were added TO acetate (1 mg, 1 eq), DIPEA (14 uL, 28 eq) and HBTU (15 mg, 14 eq) after which the solution was stirred for 15 min. A suitable amine coupling partner (4 eq) was added and the solution was stirred for another 15 min after which the reaction mixture was lyophilised, redissolved in MeCN and purified by HPLC.
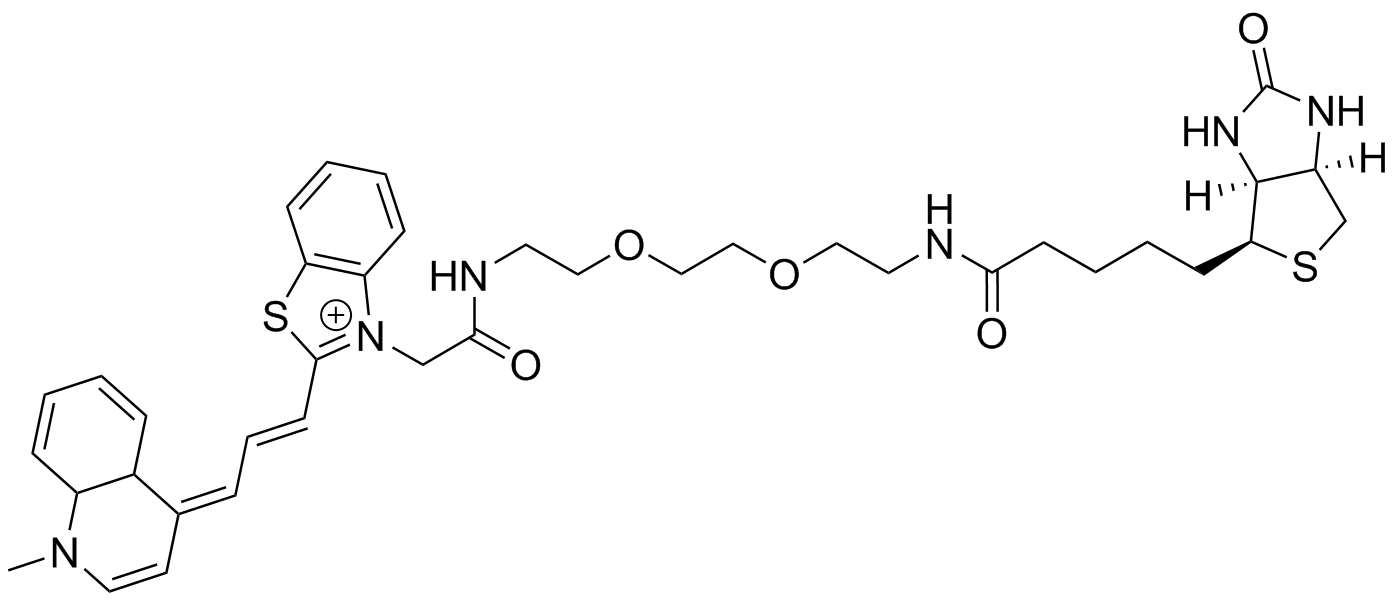
TO3 PEG2 link amine Biotin: HPLC gradient from 100% 50 mM NH4-acetate/0% MeCN to 33% NH4-acetate/67% MeCN over 45 min. HRMS (ESI) Exact mass calculated for [M+,C38H51N6O6S2+] requires m/z 731.3044 found 731.3047.HPLC: tR= 21 min
- Paige, J. S.; Wu, K. Y.; Jaffrey, S. R. Science 2011, 333, 642–646.
- Baldridge, A.; Kowalik, J.; Tolbert, L. Synthesis (Stuttg), 2010, 14, 2424–2436.
- Lee, C.Y.; Chen, Y.-C.; Lin, H.-C.; Jhong, Y.; Chang, C.W.; Tsai, C.H.; Kao, C.L.; Chien, T.C. Tetrahedron, 2012, 68, 5898–5907.
- Troponwerke GmbH & Co. DE2659543, 1978.
- Dolgosheina, E. V; Jeng, S. C. Y.; Panchapakesan, S. S. S.; Cojocaru, R.; Chen, P. S. K.; Wilson, P. D.; Hawkins, N.; Wiggins, P. A.; Unrau, P. J. ACS Chem. Biol., 2014, 9, pp 2412–2420
- Chen, K.Y.; Cheng, Y.M.; Lai, C.H.; Hsu, C.C.; Ho, M.L.; Lee, G.H.; Chou, P.-T. J. Am. Chem. Soc., 2007, 129, 4534–4535.